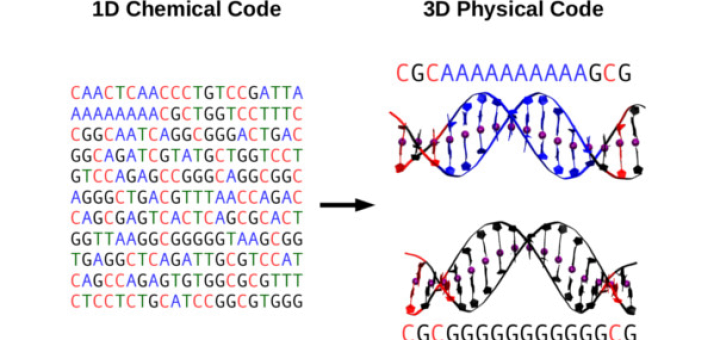
Towards Deciphering the Physical Code of DNA
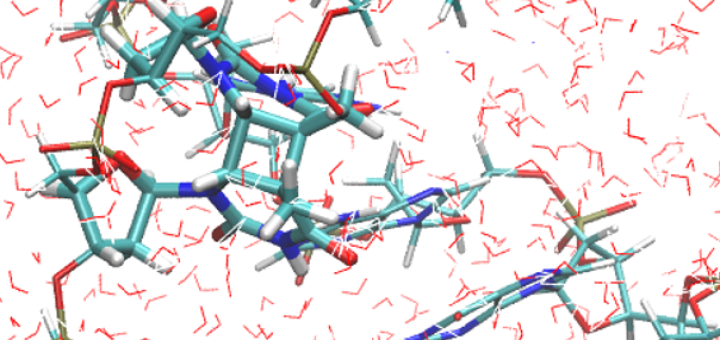
Article: published in Physical Review Letters by Rubén Pérez, IFIMAC researcher and member of the Department of Theoretical Condensed Matter Physics. The genomic material needs to be precisely organized and access to its information...