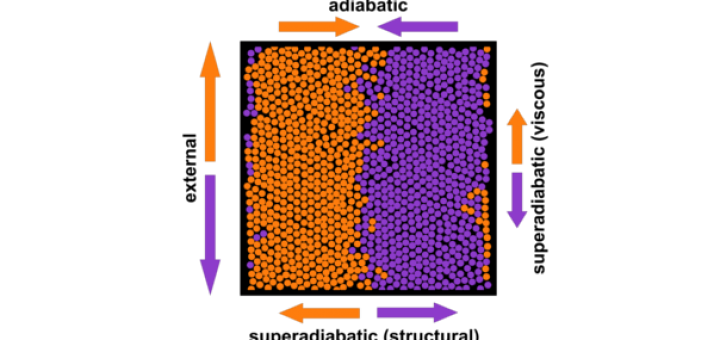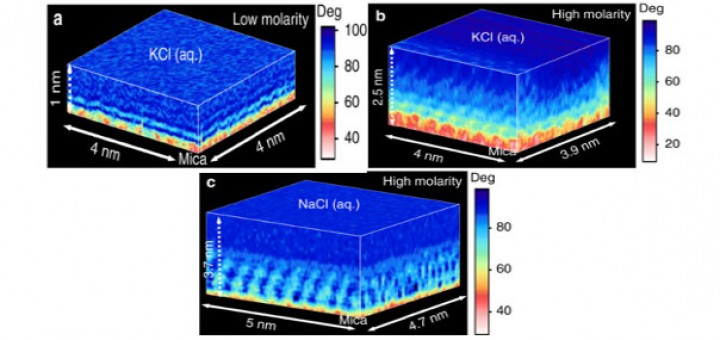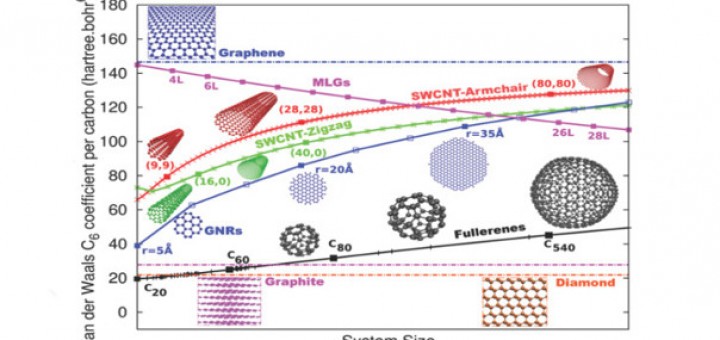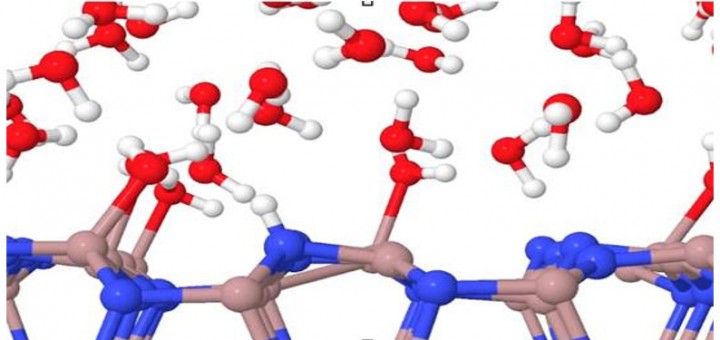
Molecular Identification and Bonding Information from High-resolution AFM
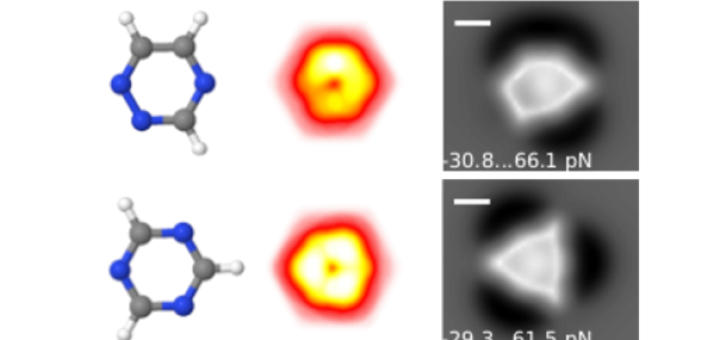
Article: published in ACS Nano by Pablo Pou and Ruben Perez, IFIMAC researchers and members of the Department of Theoretical Condensed Matter Physics. High-resolution atomic force microscopy (HR-AFM) is able to image the internal...