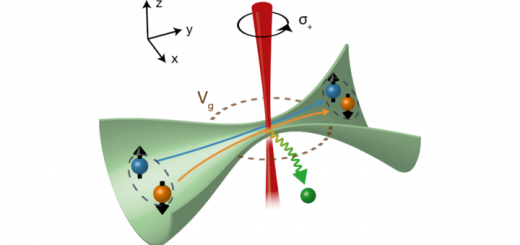
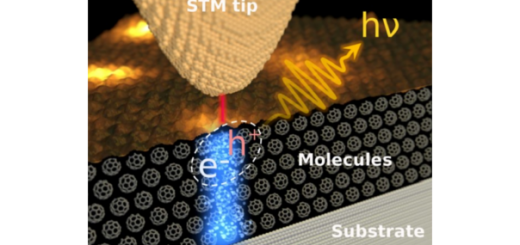
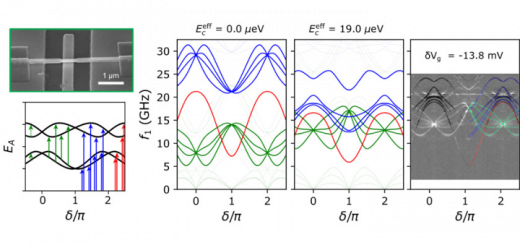Numerical evaluation of four-center molecular intergals for localized orbitals
 Wednesday, 14 January 2009, 15:0-16.00
Wednesday, 14 January 2009, 15:0-16.00
Dr. Masayuki Toyoda
Japan Advanced Institute of Science and Technology (JAIST) 1-1, Asahidai, Nomi, Ishikawa 923-1292, Japan.
ABSTRACT:
We have developed a computer routine to evaluate the four-center molecular integrals for the pseudo-atomic orbital basis sets.
This enables us to calculate the Fock exchange energy for Kohn-Sham orbitals within order-N DFT calculations.
In our test calculations with simple molecules, the convergence up to 10^-5 Hartree in energy has been successfully achieved at an acceptable computation cost.